Разоткривање скривених термодинамичких сила које покрећу хемијске реакције | Истраживања
Шта одређује како брзине реакције реагују на термодинамичке покретачке силе? И зашто се неке реакције драматично убрзавају са малим променама у покретачкој сили, док неке друге делују много мање? У нади да ће одговорити на оваква питања, теоретски хемичари у Немачкој увели су нови оквир за моделирање пејзажа реактивности хемијских реакција.
Разумевање улоге спољашњих фактора у хемијским реакцијама је централно за теоријска и експериментална истраживања хемије. Стицање дубљег увида у ове факторе може помоћи хемичарима да усмере реакције ка жељеним производима, максимизирају приносе и ограниче нежељене споредне реакције.
Хемијске реакције се могу посматрати као непрекидна потрага за стабилношћу. Из термодинамичке перспективе, ова стабилност се мери Гибсовом слободном енергијом, а реакција обично напредује дуж путање која смањује ову енергију. Међутим, овај пут није јединствен – реакције могу да еволуирају кроз више субоптималних конфигурација у зависности од спољашњих утицаја.
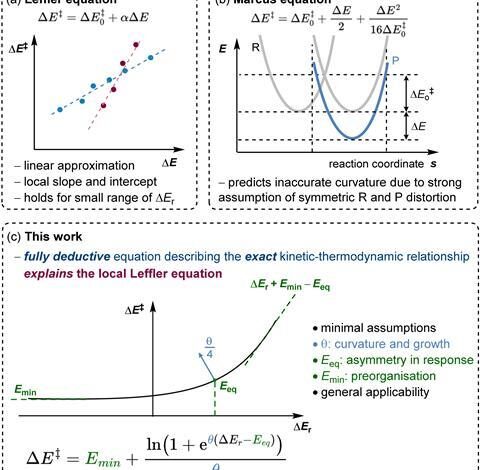
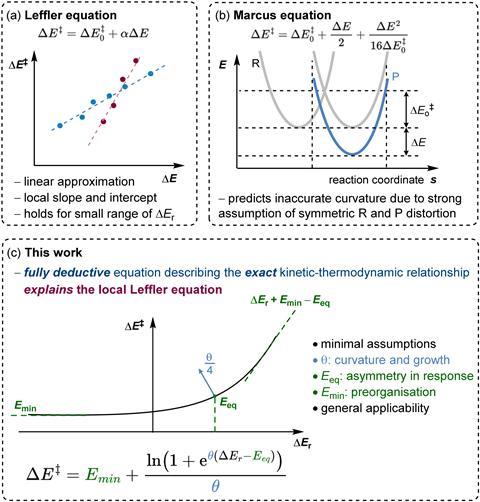
Класични модели, као што су Лефлерове или Маркусове једначине, покушали су да схвате како стопе реакције реагују на спољне покретачке силе, са циљем да идентификују омиљене путеве реакције. Међутим, ови модели су ограничени спектром термодинамичких ситуација које могу описати. Напреднији статистички приступи, иако су шири по обиму, често се ослањају на више параметара специфичних за реакцију, што може смањити њихову општу интерпретабилност.
Нова једначина са једним параметром
Сада су Едуардо Гарсија-Падиља и Гуанћи Ћиу са Института Макс Планк за истраживање угља у Немачкој предложили модел који повезује енергију активације реакције (ΔЕ‡) на његову укупну реакциону енергију (ΔЕр). Њихова једначина се ослања на један параметар – фактор закривљености θ – који одражава стабилност прелазног стања и како на њега утичу конкурентски фактори који утичу на реакцију.
За разлику од претходних приступа, модел прати кључна својства хемијских реакција без прибегавања рестриктивним претпоставкама. Резултат је оквир који одржава тачност модела заснованих на статистици уз задржавање једноставности класичних модела.
Гарсија-Падиља предвиђа да ће њихов модел помоћи хемичарима да открију нове путеве реакције откривањем скривених односа између сила које покрећу хемијске трансформације. „Неки од параметара вам говоре, на пример, да постоји минимална енергија активације која се никада неће моћи превазићи само тако што ће реакцију учинити повољнијом. Ово већ даје синтетичким хемичарима већу контролу над реакцијом од претпоставке да постоји линеарни однос слободне енергије и да ће реакција увек реаговати на исти начин.’
Гарсија-Падиља предвиђа будућност у којој ће предвиђање параметара модела за породицу реакција омогућити корисницима да предвиде „колико ће реакција одговорити и подесити одговор да повећа селективност између два могућа процеса“ под примењеном покретачком силом.
Гледајући унапред, Гарсија-Падиља мисли да ће се машинско учење појавити као природна алтернатива за предвиђање параметара модела унутар одређене породице реакција. „Ове врсте комбинација машинског учења за предвиђање не брзине реакције, већ неких физички корисних параметара биле би веома занимљиве. Чак и ако би делови машинског учења били нека врста црне кутије, добра ствар је што сами параметри модела можете лако да проверите њихову валидност.’
„(Студија) представља благовремен и утицајан допринос областима рачунарске хемије и дизајна реакција“, каже Марко Мартинез, стручњак за теоријске хемијске методе за моделирање хемијске реактивности на Универзитету МцМастер у Онтарију, Канада. „Одређивање реакционих баријера захтева оптимизацију реактаната и прелазног стања које се повезује са производом. Ово чини процес дуготрајним и веома зависним од људске стручности, па стога није погодан за приступе скрининга велике пропусности. Насупрот томе, предложени модел се може параметризовати са постојећим подацима из повезаних фамилија реакција. Ово га чини погодним за скрининг велике пропусности, нудећи најбоље од оба света: ниске трошкове рачунара и физички значајна предвиђања, омогућавајући да се боље идентификује покретачка снага која стоји иза кинетике реакције.’
Мартинез каже да модел захтева додатну анализу и валидацију. „Параметри модела су повезани са хемијски интерпретабилним концептима без дефинитивног односа између њих и хемијских својстава реактаната и производа“, објашњава он. „Поред тога, перформансе модела треба да се процене за високо асинхроне реакције или у случајевима са вишеструким конкурентским реакционим путевима са сличним баријерама.“